Komórki wchodzą w skład wszystkich organizmów roślinnych i zwierzęcych (komórka = cellula). W latach 1838-39 dwaj niemieccy badacze: botanik — Schleiden i zoolog — Schwann sformułowali tzw. komórkową teorię budowy organizmów, zgodnie z którą wszystkie istoty żywe zbudowane są z komórek. W 1846 r. czeski badacz — Jan Purkyne stwierdził, że najistotniejszym składnikiem komórki jest wypełniająca ją galaretowata, żywa substancja — plazma komórkowa, czyli protoplazma. Nikt wówczas nawet nie zakładał możliwości istnienia bytów żywych nie posiadających budowy komórkowej, dopiero z czasem rzeczywistość wprowadziła zamęt w biologicznych koncepcjach życia — odkryto wirusy. [ 1 ] Natura nie powiedziała wówczas ostatniego słowa...
Nauka niechętnie rezygnuje z raz przyjętych, powszechnie akceptowanych idei, praw. Wbrew pozorom jest skostniała, opiera się zmianom. W swych podstawach świat nauki przejawia niezmiennie skłonność do ignorancji; jakże często funkcjonuje niczym zrzędzący starzec. Przykładem jest genetyczny dogmat biologii molekularnej dotyczący etapów naturalnej ekspresji genu [ 2 ], tudzież cech niezbędnych organizmom żywym lub wykazującym w warunkach sprzyjających przynajmniej niektóre z tych cech (klasycznym przykładem są tu wirusy). Francis Crick, współodkrywca struktury molekularnej kwasu DNA, twierdził: „DNA tworzy RNA; RNA tworzy białko; proces ten jest jednokierunkowy, przynajmniej w odniesieniu do białka.". Pisał (1970 r.): „W myśl tego dogmatu trzy rodzaje transformacji są niemożliwe: białko nigdy nie tworzy białka, białko nigdy nie tworzy RNA, białko nigdy nie tworzy DNA". [ 3 ] Słuszności tego dogmatu nigdy nie dowiedziono i wielki uczony doskonale wiedział o tej groźnej słabości. Dopuszczał możliwość istnienia wyjątków, kierując uwagę badaczy właśnie na czynnik powodujący chorobę scrapie.
Dotychczas uważano, iż każdy organizm posiada kwas nukleinowy DNA, niezbędny do przechowywania i przekazywania konkretnej informacji genetycznej. Przecież nawet wirus — tzw. nukleokapsyd, gdyż składa się on właśnie z kwasu nukleinowego, ochronnego płaszcza białkowego [ 4 ], rzadko lipidów — posiada ten organiczny nośnik swoistej dlań informacji genetycznej, umożliwiający mu ingerencję w metabolizm obcej komórki, a przez to namnożenie się. [ 5 ] Kolejne przemiany prowadzące do wytworzenia białka to replikacja (powielenie cząstki macierzystej DNA), transkrypcja (uzyskanie mRNA na matrycy DNA) oraz translacja (biosynteza w rybosomach/polisomach z udziałem tRNA).
Tymczasem okazało się, że istnieją cząstki o cechach wyjątkowych, które mimo braku nawet śladowych ilości jakiegokolwiek nośnika genów [ 6 ] potrafią działać w sposób nasuwający analogie z wirusami i tzw. białkami opiekuńczymi. [ 7 ] Mowa o prionach...
Nazwę prion — tzw. przesączalny czynnik zakaźny — stworzył ich odkrywca (S. B. Prusiner [ 8 ]) od słów proteinaceous infectious particie (PrP) w źródłach polskich stosuje się zwrot „kodowany genetycznie czynnik białkowy". Do niedawna przypuszczano że prionami mogą być:
— niskocząsteczkowe wirusy posiadające otoczkę białkową; miały one odblokowywać
gen odpowiedzialny za syntezę amyloidu;
— niekodujące krótkie odcinki RNA.
Zgodnie z przeważającą obecnie teorią prion jest agregatem jednego białka komórkowego; jest to czyste białko (oligomer) obecne zawsze w komórkach w postaci tzw. prionu komórkowego (PrPc ; PrP), niezbędnego do prawidłowego funkcjonowania komórki. [ 9 ] Białka prionowe są wszechobecne w świecie zwierząt, stanowią normalny składnik organizmu, lecz ich rola wciąż jest dyskusyjna, nie została określona jednoznacznie. [ 10 ] Gen kodujący — PRNP — znajduje się u człowieka w obrębie chromosomu 20; gen ów tworzy jeden ekson.
Warto zaznaczyć, że z ustaleniami (hipotezami?) Stanleya Prusinera co do natury prionów wciąż nie zgadza się liczne grono naukowców. Dopuszczają oni możliwość, że priony mogą być prostymi wirusami zakładając, że odkrycie związanych z nimi cząstek kwasów nukleinowych pozostaje kwestią czasu… Ujmując kwestię zasadniczo, według przeciwników teorii prionu właściwy czynnik infekcyjny odpowiedzialny za gąbczaste encefalopatie nie został jeszcze wyizolowany [ 11 ]! Zresztą nawet sam Prusiner B. S. asekurując się nie wyklucza, że w skład prionów wchodzi dodatkowy komponent białkowy (tzw. białko X), dotychczas nie odkryty. Może to być także oligonukleotyd — krótki odcinek DNA otoczony białkiem prionowym (wg Liberski P.).
Przykłady konkurencyjnych poglądów:
-
D.C. Gajdusek — zakaźna jest nie pojedyncza cząstka, lecz jej agregat,
kryształ. Wpierw PrPSc tworzy jądro krystalizacji, wokół którego
odkładają się kolejne cząstki. Podobnie jak w przypadku płatków śniegu tak i tu
jedna cząsteczka może tworzyć odmienne kryształy np. szczepów scrapie;
-
Richard H. Kimberlin — autor hipotezy „wirino" według której wirus
scrapie jest hybrydą (chimerą) molekularną składającą się z cząstki kwasu
nukleinowego rozmiarów zbliżonych do wiroidu oraz płaszcza białkowego, którym
może być właśnie PrPSc;
-
Heino Diringer (Robert Koch Institut, Berlin) — obserwował cząstki
długości 10 nm, mogące być dotychczas niewyizolowanym wirusem (nieco większe
cząstki — ok. 30 nm średnicy — wykazano w mózgach chorych na CJD).
Ostateczne udowodnienie klasycznej teorii prionu wymaga konstrukcji tego białka w warunkach eksperymentalnych. Podjęte próby nie udały się lub też uzyskane wyniki są dyskusyjne, podawane w wątpliwość (np. eksperymenty na transgenicznych myszach). W warunkach in vitro doprowadzono do przekazania na prawidłowe białko wadliwej konformacji, lecz nie produkuje ono infekcyjności, tzn. w testach wykazują właściwości białka zakaźnego nie są jednak zakaźne w próbach ze zwierzętami.
Białka prionowe występują w błonach komórkowych i krążą wraz z fragmentami błony zewnętrznej i wewnętrznych pęcherzyków komórki (endosomów). Właśnie ze względu na ich stałą obecność w błonach komórkowych, oraz z powodu przyłączonych części cukrowych, początkowo sądzono, że białko PrP jest receptorem dla hipotetycznych wirusów scrapie (miały one wywoływać chorobę owiec). Istotne, że wirusa takiego nigdy nie odnaleziono; obecnie wiemy zresztą, że scrapie z wirusami nie ma żadnego związku.
W badaniach poszukujących specyficznej roli prionów w zdrowym organizmie stworzono myszy transgeniczne, które nie posiadały genów kodujących te białka i co za tym idzie, były ich pozbawione. Nie zaobserwowano u tych zwierząt większych zaburzeń, więc nie udowodniono, czy priony rzeczywiście są konieczne do życia. U myszy z uszkodzonym genem na PrP obserwuje się m. in. zaburzenia snu i czuwania, zaburzenia transdukcji sygnału (transmisji sygnału na powierzchni komórki). Doniesienia te są jednak pojedyncze, nie wszystkie wzajemnie się potwierdzają. Podaje się też, że brak prionów powoduje przedwczesne obumieranie komórek mózgowych, a ostatnie badania sugerują, że zdrowe priony chronią neurony przed samobójczą śmiercią (apoptozą). Uważa się też, że mają jakiś związek ze stresem oksydacyjnym komórki (określa się tak stan braku równowagi pomiędzy działaniem reaktywnych form tlenu a biologiczną zdolnością do szybkiej detoksykacji reaktywnych produktów pośrednich lub naprawy wyrządzonych szkód). Przypuszczalnie funkcje biologiczne PrP obejmują: homeostazę wapnia, regulację presynaptycznych stężeń miedzi, aktywność i lokalizację nauronalnej syntezy tlenku azotu, adhezję komórek. [ 12 ]
Więcej wiadomo o modyfikacjach posttranslacyjnych prionów. Do białka PrP przyłączane zostają części cukrowe oraz lipidowe (dzięki nim zakotwicza się w błonie komórkowej od strony cytoplazmy).
W komórkach może również wystąpić druga izoforma prionu (alternatywna struktura przestrzenna), jest ona jednak infekcyjna („scrapie") — ozn. PrPSc (zamienne symbole: PrPres, PrPd, Sc). Izoforma ta jest przyczyną chorób pionowych.
Wcześniej uważano, że różne formy przestrzenne białka prionowego są wynikiem alternatywnego składania eksonów — zjawiska spotykanego często, szczególnie u ssaków. [ 13 ] Okazało się, że sekwencje aminokwasów obu form białka PrP są identyczne. Było to pierwsze białko, dla którego zaobserwowano takie zjawisko. Jest to zarazem jedna z najważniejszych cech prionów.
Wspomniana forma infekcyjna („scrapie") charakteryzuje się obecnością rozbudowanej, w porównaniu do struktury PrPc, β-kartką oraz odznacza się mniejszą zawartością α-helisków. Nie są to jedyne różnice. Struktury te cechuje także różna rozpuszczalność w wodzie i podatność na trawienie enzymami proteolitycznymi (tzw. proteazy; dokonują hydrolizy wiązań peptydowych). Forma PrPSc wytrąca się w środowisku wodnym i jest odporna na proteolizę. Ma to poważne konsekwencje w patogenezie chorób prionowych.
Tabela [ 14 ]
| Forma PrPc | Forma infekcyjna PrPSc | |
|
Rozpuszczalne w środowisku wodnym |
Tak | Nie |
| Podatne na trawienie proteazami | Tak | Nie |
| Dominująca struktura drugorzędowa | α-heliks | β -kartka |
Istnieje cała grupa chorób układu nerwowego, w których czynnikiem sprawczym są tzw. patologiczne konformery (białka o nieprawidłowej konformacji); nieoficjalnie używa się niekiedy przenośni „nowotwory białek". Białka mające właściwość zmiany kształtu innych białek nazywane są chaperonowymi. Prion nie jest więc wyjątkiem, jego specyfiką jest fakt, iż zmieniając swą konformację staje się zakaźny!
Rozwiązanie struktury PrP nastąpiło głównie dzięki technice spektroskopii rezonansu magnetycznego. Natomiast w przypadku formy patogennej nie możemy uzyskać jej roztworu, ani kryształu przez co spektroskopia, krystalografia stają się bezużyteczne; dowodów pośrednich dostarczają inne metody, np. diploizmu kołowego. Cząstka prionu składa się z około tysiąca monomerów, jeśli rzeczywiście zawiera jakiś odcinek kwasu nukleinowego to ma długość on nie więcej niż 200 nukleotydów. [ 15 ]
Aby w pełni zrozumieć istotę prionów przybliżę wpierw strukturę białek.
Struktura białka:
- I-rzędowa — in. pierwotna; łańcuch polipeptydowy zsyntetyzowany przez rybosom w procesie translacji (biosyntezy); sekwencja aminokwasów w łańcuchu białkowym. Struktura ta zapisana jest w genie kodującym układ aminokwasów danego białka, a utrzymują ją kowalencyjne wiązania peptydowe [ 16 ];
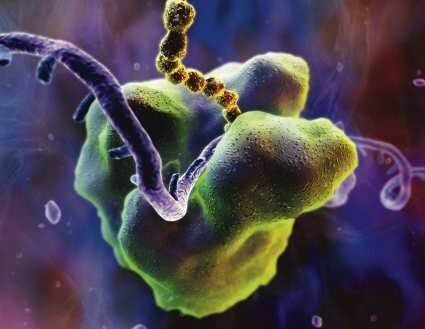 [ 17 ]
[ 17 ]Rybosom — organellum w którym powstają białka
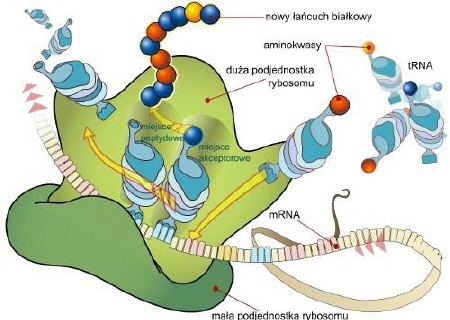
Mechanizm działania rybosomu
(schemat ukazuje translację mRNA; fazę elongacji) [ 18 ]
- II-rzędowa — ułożenie poszczególnych fragmentów
w przestrzeni, utrzymywana przez wiązania wodorowe między aminokwasami. Najbardziej stabilne układy przestrzenne (tzw. układy regularne) to alfa helisa (a-helisa) i kartka beta (struktura b, zwana też harmonijkową).
| Nazwa struktury | Wiązania |
| a-helisa | Wiązania wodorowe między grupami =C=O jednego aminokwasu, a grupą =N-H innego, odległego o 4 jednostki aminokwasowe; struktura ta jest częsta w białkach globularnych /kulistych/, np. w hemoglobinie, mioglobinie [ 19 ]. |
| Kartka b | Wiązania wodorowe między równoległymi łańcuchami polipeptydowymi; struktura ta umożliwia np. zmianę kierunku łańcucha w przestrzeni, tzw. „zakręty". Tworzone przez nią białka są zwarte, nierozciągliwe i nierozpuszczalne w wodzie, znaczny udział tej struktury stwierdzono m.in. w fibroinie jedwabiu. |
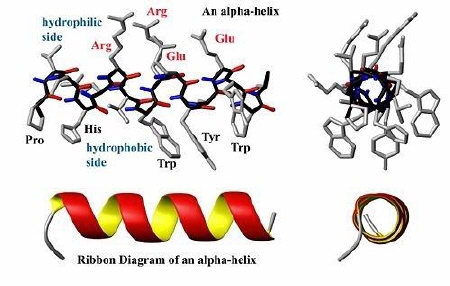
Struktura a-helisa [ 20 ]
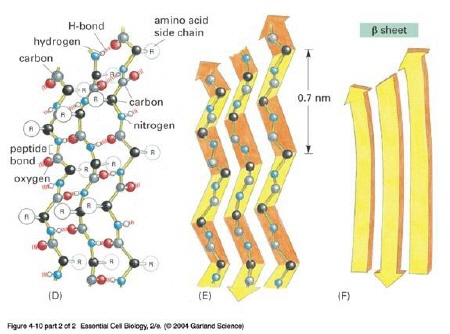
Struktura kartka b [ 21 ]
- III-rzędowa — in. konformacja — przestrzenne pofałdowanie łańcucha struktury a-helisu wynikające z oddziaływań między resztami R niektórych aminokwasów (konkretnie mogą to być wiązania jonowe, oddziaływania hydrofobowe, mosteczki dwusiarczkowe powstające między parą cystein w tej samej cząstce białka Cys-Cys, czy oddziaływania van der Waalsa). [ 22 ]
- IV-rzędowa — dotyczy tylko białek budowanych przez kilka dużych podjednostek (np. hemoglobina), łańcuchy polipeptydowe łączą się w jedną, funkcjonalną całość (in. łańcuchy o strukturze III-rzędowej tworzą układ przestrzenny).
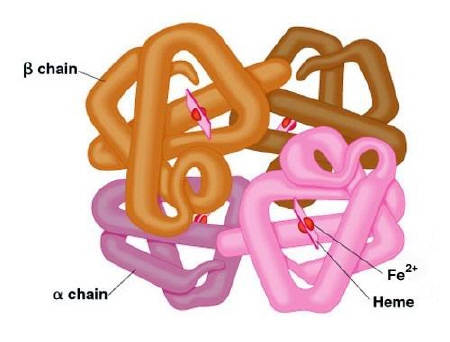 [ 23 ]
[ 23 ]Struktura hemoglobiny
Przyjmuje się, że prion nabiera cech patogennych w wyniku nagłej zmiany struktury trzeciorzędowej (łańcuch polipeptydowy); przy czym nie dotyczy to sekwencji aminokwasowej, przeobrażeniu ulega przestrzenna forma cząsteczki, stając się tzw. PrPSc -prionem powodującym scrapie(in. skarpie), chorobę występującą u kóz i owiec.
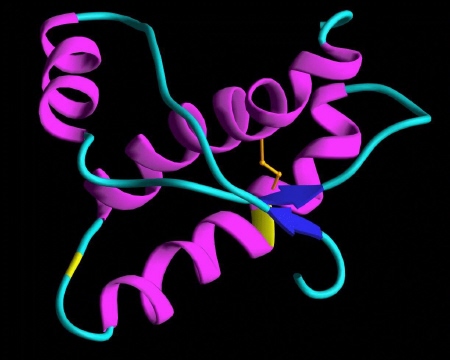
Konformacja białka PrP [ 24 ]
Konwersja PrPc do PrPSc następuje po ponownym wejściu cząsteczki do komórki poprzez jamkowe domeny lub wyłożone latryną zagłębienia. Tak więc prion może być izoformą normalnego białka lub produktem zmutowanego genu, aczkolwiek mechanizm powstawania formy PrPSc wymaga jeszcze wielu badań. Tak naprawdę wciąż nie wiemy jak to się odbywa. Zaproponowano model tłumaczący to zjawisko. [ 25 ] Otóż białko PrP przyjmuje dwie formy przestrzenne, których zmiana jest procesem spontanicznym. Forma PrPc nie posiada zdolności tworzenia „polimerów" w przeciwieństwie do formy infekcyjnej, która przy udziale β-kartki może przyłączać kolejne cząsteczki tego białka. Proces ten jednak jest całkowicie odwracalny do momentu osiągnięcia przez polimeryzujące molekuły PrPSc rozmiaru krytycznego. Osiągnięcie takiego rozmiaru oznacza, że polimer cząsteczek PrPSc nie może już się zmniejszyć, nie ma wówczas możliwości powrotu do konformacji PrPc. W ten sposób powstają tzw. złogi β-amyloidowe (beta-amyloidowe), stanowiące według wielu badaczy przyczynę m.in. choroby Creutzfeldta-Jakoba (CJD). [ 26 ] Złogi te niekiedy pękają i są przekazywane do komórek potomnych przy podziale komórkowym. [ 27 ] Tak więc reasumując, mechanizm szkodliwości prionów zakaźnych (neurotoksyczność) polega na gromadzeniu się ich w komórkach, w błonach wakuol, zaburzeniu recyrkulacji błon oraz akumulowaniu zewnątrzkomórkowo w postaci tzw. beta-amyloidu. Przypuszczalnie przyczyniają się do indukcji apoptozy neuronów, powodują zaburzenia glikozylacji i metabolizmu PrP.
Naukowcy stwierdzili że priony są białkami, które wnikając do komórki indukują syntezę potranskrypcyjnych czynników modyfikujących, w wyniku czego pojawia się zmodyfikowane białko.
Możliwy schemat przemian:
1.
Białko PrP o konformacji prawidłowej (globularnej).
2. W wyniku np. mutacji punktowej dochodzi do zamiany aminokwasów,
konkretniew pozycji 102 leucyny na prolinę.
3.
Powstaje wariant nieprawidłowy konformacji; płat/kartka o strukturze
beta.
Przybranie innej formy przestrzennej przez białko prionowe jest jednoznaczne z nabyciem przez niego właściwości białka infekcyjnego (patogennego) [ 28 ]! Znamienne, że wciąż nie znane są przyczyny zmian PrP w postaci patogennej.
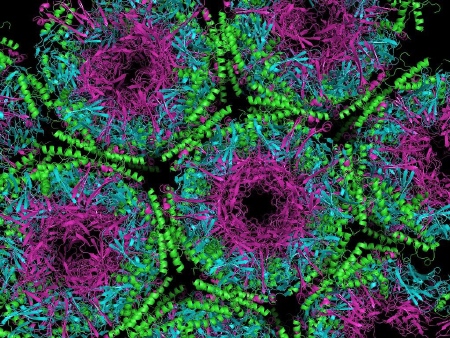
Struktura krystalograficzna białka prionowego [ 29 ]
Zmiana kształtu jest przekazywana kolejnym cząstkom białka. Specyfika polega na tym, że nie jest to typowe powielanie, namnożenie, lecz przekazanie zmian czysto strukturalnych innym, występującym już na obszarze komórki cząstkom pokrewnym, zostają one niejako zmuszone do przyjęcia nieprawidłowej konformacji przestrzennej. Zmienione priony są odporne na działanie enzymów (proteaz), nie mogą zostać strawione, akumulują się więc wewnątrz neuronów (tworzą płytki amyloidu, tzw. pałeczki prionowe [ 30 ]) i zlepiają w charakterystyczne dla poszczególnych szczepów złogi, uszkadzając ciało komórki (wakuolizacja) oraz nabierając cech neurotoksycznych. Konsekwencją są ubytki tkanki mózgowej, stąd też nazwa gąbczasta encefalopatia. Postępująca degeneracja powoduje ciężkie zaburzenia psychiczno-ruchowe, jak dotąd nieuchronnie prowadząc do śmierci. Obecność patogennych prionów, które są wszak w normalnej postaci cząstkami słabo immunogennymi, nie powoduje wytworzenia wykrywalnych ilości przeciwciał we krwi, czy płynie mózgowo-rdzeniowym (stąd trudności w diagnostyce), są one jednak komercyjnie dostępne (wytworzone laboratoryjnie). Wszystkie choroby prionowe ludzi i zwierząt charakteryzują się zmianami zwyrodnieniowymi układu nerwowego, dlatego określane są wspólnym skrótem TSE (transmissible neurodegenerative disease = pasażowalne encefalopatie gąbczaste).
Przyjęto, że choroby te charakteryzują się gąbczastą patologią oraz obecnością zgrupowań kryształów PrP (widoczne pod mikroskopem elektronowym jako skręcone włókienka w homogenizowanym preparacie tkanki mózgowej) zwanych SAF (scrapie associated fibrils).
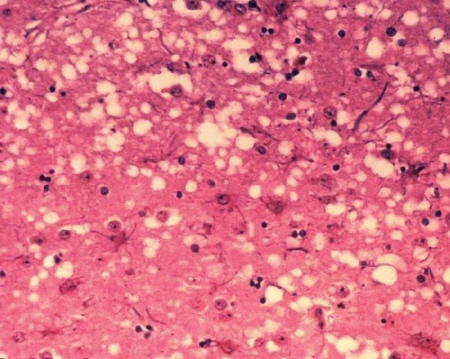
Mikrofotografia tkanki mózgowej ujawniająca zmiany cytoarchitektoniczne
w gąbczastej encefalopatii bydła. Obecność wakuol („dziur") w istocie szarej
daje gąbczasty wygląd tkanki [ 31 ]
W przeciwieństwie do innych czynników patogennych jak bakterie, wirusy, priony nie są obce względem atakowanego organizmu, lecz stanowią jego naturalne elementy strukturalne! Zmiana fenotypu związana z prionami coraz częściej uznawana jest za formę informacji genetycznej, mimo, że w jej przekazywaniu nie pośredniczą kwasy nukleinowe (dotyczy jedynie organizmów haploidalnych, rozmnażających się przez podział). Z tego punktu widzenia badacze zaliczają ją do zjawiska epigenetycznego, podobnie jak tzw. kod histonowy, czyli modyfikacje posttranslacyjne histonów powodujących rozmaite zmiany nie tylko w stanie chromatyny, a także komórki. [ 32 ]
Przy badaniach związanych z prionami obowiązują precyzyjnie wyznaczone procedury: poziom bezpieczeństwa P2 (śluza, fartuch, czapka itp.) do P3 (dodatkowo tworzy się warunki ujemnego ciśnienia).
Cząstki patogennych prionów są zdumiewająco odporne, w warunkach tzw. suchego spalania ulegają zniszczeniu dopiero w temperaturze +360ºC [ 33 ] lub, aż +600ºC. [ 34 ] Przy podwyższonym ciśnieniu do spalenia wystarcza temperatura około +135ºC. [ 35 ] Czynnik scrapie badał (lata 1939-1953) D. R. Wilson, jeden z jego kolegów pisał: „(...) czynnik chorobotwórczy jest niezwykle odporny na wszelkie substancje niszczące. Przetrzymał półgodzinne gotowanie. W zamrażarce przetrwał dwa miesiące. Był nieczuły na środki dezynfekcyjne, zawierające silne stężenie formaldehydu, fenolu i chloroformu. Przenikał przez najgęstsze filtry i pozostał w roztworze umieszczonym w wirówce przy czterech tysiącach obrotów na minutę. Mógł przeżyć w wysuszonym mózgu przez co najmniej dwa lata, był odporny na silną dawkę promieniowania ultrafioletowego. Przenosił się z jednej owcy na drugą wstrzyknięty podskórnie, domięśniowo czy bezpośrednio do mózgu." [ 36 ]
Naukowcy postanowili sprawdzić jak priony działają na białko obce naturze, stworzone przez człowieka, w tym przypadku połączono białka dwóch drożdży: "SC" (Saccharomyces cerevisiae) i "CA" (Candida albicans). Wynik: białko okazało się podatne zarówno na działanie oryginalnych prionów "SC", jak i "CA", tym samym właściwie było podatne na działanie prionów obcych sobie gatunków. Możliwe więc iż podatna na zmiany nie musi być cała cząsteczka, lecz wystarczy by jej fragment był wrażliwy na czynnik chorobotwórczy, a dojdzie do przeniesienia choroby.
Do dziś poznano następujące choroby prionowe człowieka, wszystkie są neurodegeneracyjne, powstają w obrębie ośrodkowego układu nerwowego. Wszystkie również — jak dotąd — zawsze kończą się śmiercią, leczenie jest wyłącznie objawowe! Nie jest znane lekarstwo na choroby prionowe.
Choroby prionowe człowieka:
KURU
Chociaż dzieje ludzkich chorób prionowych zaczęły się właśnie od niej dla przeciętnego człowieka choroba ta pozostaje właściwie nieznana. Wynika to po części z faktu, iż ma (miała) charakter lokalny, występując wyłącznie w obrębie plemienia Fore, mieszkańców górzystych rejonów Papui-Nowej Gwinei („kuru" oznacza w języku Fore „drżeć z zimna/gorączki"). Praktykowali oni rytualny kanibalizm w dowód szacunku dla zmarłych. [ 37 ]
Okres zwiastunów (prodromalny) — trwa od pojawienia się pierwszych objawów do pełnego rozwoju objawów klinicznych — obejmuje: bóle głowy, brzucha i kończyn (stawów), utratę masy ciała.
Objawy (występowały po okresie kilkunastu miesięcy lub nawet 40 lat):
1. Zawsze występuje śmiertelna ataksja móżdżkowa [ 38 ], której towarzyszą drżenie, ruchy mimowolne o typie pląsawiczych/atetotycznych oraz nietrzymanie moczu i kału.
2. Kuru ma trzy fazy:
— chory jeszcze chodzi — chód staje się niepewny, przechodzi w nasiloną ataksję i abazję [ 39 ]. Pojawia się delikatne „drżenie z zimna", potem chwianie, próby utrzymania równowagi (kurczowe przytrzymywanie się gruntu za pomocą szponowato zgiętych palców stóp). Może pojawić się poziomy oczopląs. Odruch podeszwowy zawsze zgięciowy. [ 40 ] Zawsze obecny klonus (trząs) rzepki, ewentualnie klonus stopy, czyli ciąg mimowolnych skurczów włókien mięśniowych wywołanych przez nagłe rozciąganie mięśnia.
— chory może siedzieć — zaczyna się, gdy chory nie jest w stanie chodzić bez trwałego podparcia (koniec przychodzi z chwilą, gdy nie potrafi siedzieć bez podparcia). Postępowanie objawów: niestabilność postawy, nasilona ataksja, grubofaliste drżenie, dyzartria (jeden z typów zaburzeń mowy, wynikający z dysfunkcji aparatu wykonawczego: języka, podniebienia, gardła, krtani).
— terminalna — osoba jest już całkowicie unieruchomiona. Nie trzyma moczu i kału. Pojawia się ciężka dysfazja (zaburzenia mowy, brak koordynacji słów i trudności ułożenia ich w zdania mimo sprawności odpowiednich mięśni). Specyficzny dla kuru jest brak otępienia (lub też pojawia się ono w bardzo zaawansowanych stadiach). Występują prymitywne odruchy: ssania, gryzienia, chwytny. Zmiany nastroju, od euforii do depresji. Następnie przymusowy płacz i śmiech (tzw. „patologiczny śmiech", stąd choroba zwana jest „śmiejącą się śmiercią"), otępienie (demencja). Ostatecznie wyniszczeni i niezdolni do ruchu chorzy oczekiwali śmierci głodowej lub ginęli wskutek zachłystowego zapalenia płuc, jeżeli podano im odrobinę wody. Choroba zaczęła zanikać od lat 40. XX w. kiedy to rząd ostatecznie wyegzekwował zakaz rytualnego spożywania mózgów zmarłych współplemieńców. Obecnie kuru nie występuje. [ 41 ]
Choroba Creutzfeldta-Jakoba (CJD)
Opisana w 1921 r. przez niemieckich neurologów, byli to Hans G. Creutzfeldt i Alfons Jakob. Przeważnie występuje u osób w wieku około 60 lat, z częstością 1 przypadku/milion/rok. [ 42 ] Wyróżniono postaci: sCJD (samoistna, występuje sporadycznie), jCJD (przepasażowna/jatrogenna), fCJD (rodzinna), vCJD (wariant CJD). Istotną cechą tej choroby jest to, że nie pojawia się skierowana przeciwko patogenom reakcja immunologiczna, bowiem układ odpornościowy nie rozpoznaje czynnika odpowiedzialnego za chorobę. Wynika to z faktu, że patogenem jest białko, naturalnie występujące w organizmie ludzkim.
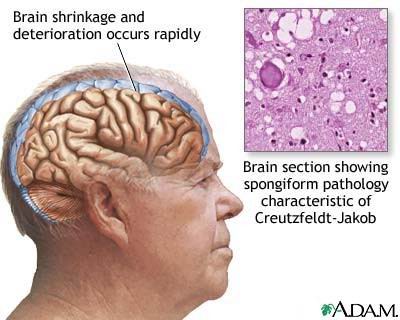 [ 43 ]
[ 43 ]CJD
W obrazie klinicznym w blisko 70 procentach przypadków występuje triada objawów: postępujące otępienie psychiczne, mioklonie [ 44 ], typowy zapis EEG. U około 30 procent chorych rejestruje się objawy prodromalne: osłabienie, zaburzenia snu (bezsenność, rzadziej nadmierna senność) i jedzenia. W co piątym przypadku choroba zaczyna się nagle. U 2/3 pacjentów występują zaburzenia zachowania i deterioracja intelektualna (pogorszenie się, psucie się, utrata wartości, spadek jakości). Także u 2/3 obecne są zaburzenia neurologiczne (chodu, podwójne widzenie bądź utrata wzroku, hemianopsja (połowiczna ślepota, ograniczenie pola widzenia do jednej połowy).
Inne objawy: zaburzenia wyższych czynności nerwowych, palinopsje [ 45 ], mimowolne ruchy choreoataktyczne, objawy czuciowe potem zmiany neurologiczne (sztywność mięśniowa, drżenia, porażenie nerwów czaszkowych), po roku (u 90 procent przypadków), dwóch (niekiedy upływa dłuższy okres) następuje śpiączka i śmierć. [ 46 ]
U 10-15 procent choroba ma charakter dziedziczny, jest wynikiem mutacji genu kodującego białko pionu. {P:47|Encyklopedia Multimedialna PWN' 2000 podaje jeszcze, że CJD wywoływana jest przez tzw. wirusy powolne, charakteryzuje ją bardzo długi okres inkubacji, do 30(40) lat (na pewno więcej niż 10 lat).} Patogenne priony obecne są w kępkach Periera jelita i wyrostka robaczkowego [ 48 ], migdałkach [ 49 ], układzie chłonnym/limfatycznym wraz z węzłami, śledzionie, płucach i sercu.
Źródła nieumyślnych zakażeń: transplantacja rogówki (na świecie do 2000 r. odnotowano trzy przypadki), implantacja opony twardej i elektrod do mózgu (około stu przypadków), podskórne podanie hormonu wzrostu [ 50 ], badania endoskopowe, przeszczep wątroby lub błony bębenkowej, nieprawidłowo odkażone narzędzia chirurgiczne, uszkodzenia powłok ciała (rzeźnicy i in.). Badania eksperymentalne wskazują na możliwość zarażenia poprzez transfuzję zakażonej krwi. Nie jest znana dawka zakażająca [ 51 ] i wielkość populacji zakażonej.
Ostatnimi czasy pojawiła się kontrowersyjna kwestia nowego wariantu, tzw. vCJD (przedstarcze otępienie umysłowe); został zidentyfikowany w roku 1996. Jest wynikiem przepasażowania encefalopatii gąbczastej bydła (BSE) na człowieka. Przy tym wariancie średni wiek zachorowania to 26 lat (przedział 19-41 lat); vCJD rozwija się do roku (7,5-23 mies.). Obraz kliniczny: zaburzenia zachowania (lęk, agresja) jako objawy prodromalne, ciągłe dyzestezje i ból w stopach [ 52 ], ataksja (wcześnie), otępienie (późno). Brak typowego EEG.
Prawdopodobnie upośledzenie mowy przy kuru i CJD nie wynika z bezpośrednich uszkodzeń wyższych ośrodków mowy w mózgu, lecz jest skutkiem zaburzenia funkcji móżdżku, zwłaszcza synchronizacji czynności aparatu oddechowego, krtani, podniebienia, języka i ust, niezbędnej do prawidłowej fonacji. Upośledzeniu ulegają też działania móżdżku związane z zachowaniem równowagi.
Syndrom Gertsmanna-Strausslera-Scheinkera (GSS)
Objawy: ataksja oraz inne symptomy wskazujące na uszkodzenie móżdżku.
Śmiertelna dziedziczna/rodzinna bezsenność (FFI)
Objawy: po okresowych trudnościach w zasypianiu następuje otępienie. Początek w wieku 37-61 lat, trwa średnio 13 miesięcy (7-25 miesięcy). Została opisana w przypadku jednej 6-pokoleniowej rodziny. FFI występuje w równych proporcjach niezależnie od płci. Obraz kliniczny: szybko postępująca bezsenność, zaburzenia autonomiczne (nadmierna potliwość, podwyższona temperatura, nadciśnienie) i endokrynologiczne (zniesienie dobowego rytmu wydzielania somatotropiny [ 53 ] oraz stale podwyższony poziom kortyzolu przy obniżonym poziomie hormonu adrenokortykotropowego [ 54 ], ataksja, mioklonie, objawy piramidowe, demencja.
Syndrom oraz bezsenność należą do chorób dziedzicznych, występują u osób w wieku średnim.
Rodzinna postępująca glioza podkorowa
Choroba charakteryzuje się nadmiernym rozrostem astrocytów — największe komórki glejowe, sieć ich wypustek stanowi zrąb dla układu nerwowego — w obrębie kory mózgowej z jednoczesną atrofią mózgu. Przyczyna choroby pozostaje nieznana. Klinicznie manifestuje się postępującym otępieniem bez istotnych zmian neurologicznych. Opisano kilka przypadków choroby o długoletnim przebiegu do 20 lat. Może przypominać inne szczególnie postępujące porażenie nadjądrowe. [ 55 ]
Zespół Alpersa — in. choroba Alpersa (Alpersa-Huttenlochera)
Rzadka, postępująca, uwarunkowana genetycznie choroba neurodegeneracyjna ośrodkowego układu nerwowego, o początku w okresie niemowlęcym lub wczesnym dzieciństwie. Może być spowodowana mutacjami w locus 15q25 w genie kodującym mitochondrialną polimerazę gamma. Dziedziczona w sposób autosomalny recesywny. Chorobę opisał jako pierwszy Alfons Maria Jakob (1884-1931), a publikacje na jej temat przedstawili trzej jego studenci: Souza, Freedomi Bernard Jacob Alpers (1900-1981). Kolejny opis zawdzięczamy duńskim neurologom — Erna Christensen i Knud Haraldsen Krabbe (1949 r.). Eponim medyczny (termin używany w medycynie) choroby Alpersa wprowadzono w 1963 roku. [ 56 ]
Choroby zwierzęce:
Scrapie (z ang. drapanie się) — in. kołowacizna, trzęsawka owiec.
Choroba znana od XVIII wieku (opisana w Szkocji), miała początkowo charakter endemiczny. Objawy: utrata koordynacji ruchowej, pobudliwość, intensywne swędzenie prowadzące do wydrapywania sobie przez zwierzę wełny lub sierści. Występuje zakażenie boczne (owce zakażają się wzajemnie, nie wiadomo jak), ponadto przekazywanie pionowe owca ----à jagnię, przypuszczalnie w trakcie porodu. [ 57 ] W latach 30. XX w. okazało się, że scrapie jest zaraźliwa — kilkunastu tysiącom owiec islandzkich podano szczepionkę uzyskaną na bazie wyciągu z mózgów owiec szkockich. W ciągu kilku lat padło 1,5 tys. zaszczepionych sztuk (stąd też miejscowy weterynarz Sigrudsson sformułował hipotezę o istnieniu powolnych chorób wirusowych, rozwijających się dopiero w kilka lat po zakażeniu). [ 58 ]
Zakaźna encefalopatia norek (TME)
Przewlekła, wyniszczająca choroba jeleni {P:59|W Wyoming (USA) wśród jeleni choroba jest bardzo silnie zakaźna; 30 procent populacji stanowej jest zakażone. Nie wiadomo jakie są potencjalne skutki tego zjawiska dla ludzi.} i antylop.
Gąbczasta encefalopatia bydła (BSE: Bovine Spongiform Encephalopathy) — inaczej choroba szalonych krów
Opisana po raz pierwszy w Wielkiej Brytanii (1986 r.) [ 60 ], kiedy to wytrzebiono ponad 150 tys. zwierząt, aby nie dopuścić do rozprzestrzenienia się choroby. [ 61 ] Już wtedy przypuszczano, pamiętając chorobę kuru, że istnieje możliwość związku między BSE, a CJD człowieka. Objawy: brak koordynacji ruchów, nieprawidłowa postawa, ataksja tylnych kończyn, drżenie mięśniowe, utrata wagi i mleczności. Zgon po okresie objawowym trwającym 2-3 tygodnie do roku. Zakażenie prawdopodobnie tylko drogą pokarmową. [ 62 ] Do niedawna uważano, iż chorobę spowodowały dodawane do pasz podroby z zarażonych owiec, obecnie część naukowców uważa, że to owce zaraziły się wtórnie od krów: scrapie nie sposób bowiem odróżnić od owczego BSE.
Dotychczas niejako „z natury" określony rodzaj prionu (np. krowi) występował wyłącznie w organizmach zwierząt jednego gatunku, przez co drogi szerzenia się choroby były mocno ograniczone. Sytuacja zmieniła się z początkiem produkcji żywności (mięsa) na skalę przemysłową, z wykorzystaniem metod tuczu, chowu obcych naturalnym uwarunkowaniom fizjologicznym zwierząt. Gwałtowna zmiana warunków (np. cieplna, chemiczna obróbka paszy), wielokrotne wtórne zakażanie zwierząt (karmienie mączką mięsno-kostną pochodzącą z różnych gatunków -przymusowy kanibalizm!), odejście przemysłu utylizacyjnego od wypłukiwania tłuszczu, obniżenie temperatury przetwarzania odpadów, rychło doprowadziły do wyhodowania prionów wyjątkowo odpornych, zdolnych do zakażania szeregu gatunków. Należy przyznać, że wszystko wskazuje na to, że właśnie człowiek stworzył białkom prionowym idealne warunki do przyspieszonej ewolucji kreując wpierw BSE, potem nowe postaci CJD. Białka prionowe potrafią pokonywać bariery międzygatunkowe!
Takiego wariantu nie przewidywali nawet najwięksi fantaści. Prawdopodobnie po dostaniu się drogą pokarmową do jelita, priony przenikają do tkanki limfatycznej przewodu pokarmowego i naczyniami limfatycznymi docierają do śledziony, węzłów chłonnych, migdałków. Tam dochodzi do ich namnożenia i przedostania się do unerwiających te narządy nerwów. Następnie poprzez włókna nerwowe priony docierają do rdzenia kręgowego i mózgu. Przypuszczalnie w ich rozprzestrzenianiu uczestniczą limfocyty B.
W marcu 1996 r. ministerstwo Wielkiej Brytanii uznało oficjalnie za wiążącą opinię naukowców wskazujących na związek między jedzeniem zakażonej wołowiny, a ludzką chorobą CJD. W tym okresie naukowcy byli niemal pewni, że jedzenie artykułów (tj. mózgu wołowego i rdzenia) od zwierząt cierpiących na BSE powoduje u człowieka nieuleczalną chorobę mózgu. Tak więc teoretycznie jeżeli do cyklu produkcyjnego dostało się zakażone mięso produkty krowie mogą przenosić na ludzi CJD. [ 63 ] Zakaźność jest tu kwestią/problemem dawki. Wiadomo, że krowę można zakazić gramem mózgu, jednak nie znamy przy tym najmniejszej wartości dawki infekcyjnej. Mimo oficjalnych zakazów bydło karmiono preparatem białkowym pochodzącym z mięsa owiec, prawdopodobnie zakażonych scarpie. Nie dowodzi to ostatecznie, że to one były przyczyną choroby krów. [ 64 ] Zastanawiające jednak, że w latach 80. XX w. zawartość preparatu mięsno-kostnego w brytyjskiej karmie wzrosła z jednego do dwunastu procent (efekt dewaluacji funta i wzrostu cen soi i preparatów rybnych). Aktualnie obowiązuje w Wielkiej Brytanii zakaz dodawania produktów pochodzenia zwierzęcego do pasz. Na granicy bada się nie zwierzęta, lecz ich certyfikaty! Przyjęto, że mięso cieląt i krów młodszych niż 30 miesięcy jest zdrowe. Komisja Europejska 1 stycznia 1998 r. zakazała sprzedaży móżdżków cielęcych, jagnięcych i kozich, rychło specjały te znikły ze stołów i restauracji UE.
Na szczęście epidemia BSE powoli wygasła: w 1994 r. obserwowano około 23 tys. przypadków, w 1995 r. tylko 14 tys.
Jeżeli żelatyna produkowana jest w warunkach eksperymentalnych z mózgu, to jest zakaźna. Cykl produkcyjny zmniejsza infekcyjność blisko 10 tys. razy — mimo to ilość pozostałych dawek infekcyjnych (w przypadku scarpie u chomików jest to wielkość rzędu 10 do potęgi 9, u krowy z BSE znacznie mniej rzędu 10 do 7) wystarcza do zakażenia około 300 tys. ludzi (teoretycznie!). [ 65 ] Narodowy Instytut Zdrowia USA /NIH/ poinformował, że eksperymenty laboratoryjne wskazują na możliwość przenoszenia BSE na drób, np. kury. Mogą być już zarażone brytyjskie świnie, więc wybicie bydła prawdopodobnie na niewiele się zda (Brown Paul). Uczeni amerykańscy wykazali także istnienie możliwości zainfekowania m.in. myszy, antylop (w 1986 r. w zoo zdechła na encefalopatię gąbczastą afrykańska antylopa nyala), tygrysów, pumy [ 66 ] oraz małp (w tym szympansów), nawet kotów domowych (w latach 1990-1994 w Wlk. Brytanii, prawdopodobnie przez spożycie zakażonej karmy, zdechły 63 koty).
Również w przypadku kuru i CJD łatwo zakazić eksperymentalnie nie tylko duże naczelne (szympansy), lecz i małpy Starego i Nowego Świata, znacznie trudniej dokonać tego u myszy (ze scrapie sytuacja jest dokładnie przeciwna: stosunkowo łatwo jest zakazić myszy czy chomiki trudniej małpy Starego i Nowego Świata, a szympansy wykazują całkowitą odporność).
Nadal nie potrafimy ustalić wiarygodnie, jakim źródłem zagrożenia dla człowieka są inne organizmy. Objawy chorób prionowych ujawniają się długo, niektóre gatunki nie zapadają na nie (nie wyklucza to jednak ich potencjalnej roli w przenoszeniu patogenu), wciąż nie znamy roli zmienności osobniczej w „wędrówce" tych białek. Nie określono dawki infekcyjnej, a w organizmach niektórych zwierząt stwierdzono obecność kilku form białka prionowego.
W mózgach niektórych ludzi i zwierząt cierpiących na choroby prionowe nie udaje się stwierdzić patologicznej odmiany białka PrPSc, część mutacji w obrębie komórkowego genu PrPc nie prowadzi bowiem do jego powstania, aczkolwiek mają one związek z występowaniem objawów neurologicznych. Wśród biochemików pojawiły się w związku z tym koncepcje, że istnieje dodatkowy, nieznany jeszcze czynnik wywołujący choroby prionowe; według Prusinera jest to inna postać prionów, tzw. CtmPrP. Białko to nie jest wydzielane poza komórkę, jego obecność ma pobudzać komórki do syntezy transbłonowej wersji białek prionowych oraz powodować niszczenie neuronów. Niewykluczone, iż komórki posiadające owe białka stają się bardziej podatne na bodźce wywołujące apoptozę — genetycznie zaprogramowane samobójstwo… Zgodnie z tymi przekonaniami niektóre mutacje genu PrPc mogą bezpośrednio przekształcać białko kodowane przez ten gen w postać CtmPrP; objawom chorobowym nie towarzyszą wówczas priony PrPSc.
Nie wszyscy są tak samo wrażliwi na choroby prionowe — podatność zależy od odcinka DNA zwanego polimorficznym kodonem 129 (ściśle: od kombinacji kodowanych przez niego aminokwasów Met i Val). Najszybciej (kilka lat od zakażenia) umierają osoby Met/Met [ 67 ], zaś przypuszczalnie najwolniej Val/Val. [ 68 ] W 1999 r. odkryto drugi gen zlokalizowany na tym samym chromosomie, posiada częściową homologię z białkiem prionu, a w pewnych warunkach (wyłącznie eksperymentalnych!) mogą powstać hybrydowe transkrypcje. Białko to nie wykazuje ekspresji w mózgu, nie stwierdzono tam jego obecności.
Prognozy:
Wiadomo, że zjedzono ponad 750 tys. sztuk bydła zarażonego BSE.
Warianty:
-
optymistyczny — epidemia BSE pociągnie za sobą śmierć kilkuset osób;
-
pesymistyczny — odpowiednio 140 tys. ofiar (w oparciu m.in. o symulacje
komputerowe przeprowadzone przez tygodnik „Nature").
Czy spełnią się katastroficzne prognozy Stephena Dellera, mikrobiologa z Burnley Hospital w północnej Anglii, głoszącego, że wkrótce czeka nas tragedia na miarę AIDS? W roku 2001 ostrzegał, że liczba Brytyjczyków zarażonych chorobą Creutzfeldta i Jakoba, pochodną od BSE, może przekraczać 2 miliony. [ 69 ] To samo miało wydarzyć się w części kontynentalnej Europy. Paolo Gulisano pisze: „Pomimo odnotowania w Wielkiej Brytanii tysięcy przypadków gąbczastej encefalopatii bydła, nie doszło do wybuchu epidemii. Dało się natomiast zauważyć społeczną psychozę w związku z możliwością przenoszenia choroby poprzez spożywanie mięsa wołowego. Do ataku epidemii nie doszło dzięki ścisłej profilaktyce i zaprowadzonym kontrolom sanitarnym oraz wyrzeczeniom smakoszy wołowych steków. Pozostaje jednak pytanie: na ile uzasadnione było podnoszenie tak głośnego alarmu?" [ 70 ]
Nie umiem udzielić wiążącej odpowiedzi. Priony stanowią realne zagrożenie, albo wymarzony środek socjotechniczny pomagający w osiągnięciu konkretnych celów w sferach polityki, biznesu, mediów. Na strachu doskonale się zarabia. Strach ma wielkie oczy, lecz jeszcze większą siłę. Szczepionki, lekarstwa, poradniki, sprzedaż rośnie. Zarazem zagrożenie jest doskonałym tematem zastępczym angażującym masy. Jeśli akurat zagrożenia nie ma — wykreuj je. Strach ukazuje prawdziwą moc mediów i pieniądza oraz ludzką hipokryzję.
Priony nasuwają mi wiele skojarzeń np. z SARS (Severe Acute Respiratory Syndrome — Zespół ostrej ciężkiej niewydolności oddechowej), chorobą która zrujnował turystykę głównie w wielu rejonach Azji. Był to rodzaj nietypowego zapalenia płuc, pojawił się w końcu 2002 r. w prowincji Guangdong, na południu Chin. Po pewnym czasie choroba rozprzestrzeniła się drogą podróży lotniczych, do chwili obecnej śmiertelność w przypadku zachorowania na SARS jest oceniana na około 4-7 procent. [ 71 ] 16 kwietnia 2003 r. dzięki globalnej współpracy 13 ośrodków naukowych z 10 krajów (m.in. z Hongkongu, Chin, Kanady, Wielkiej Brytanii, USA) potwierdzono, że przyczyną choroby SARS jest nowy koronawirus. W roku tym WHO zarejestrowała na całym świecie 2960 przypadków SARS, z czego zmarło 119 osób. „Te dane epidemiologiczne są odwrotnie proporcjonalne do skali zainteresowania w mediach, jaki miała ta epidemia. Widać to przede wszystkim, jeśli porównamy stopień śmiertelności tej choroby z poziomem umieralności na skutek innych chorób zakaźnych (...). O SARS pisało się na pierwszych stronach gazet, informacje o nim pojawiały się jako pierwsze w telewizyjnych wiadomościach, aż do momentu, kiedy WHO ogłosiła, że na całym świecie choroba ta została powstrzymana. Nie oznacza to jednak, że zniknęła albo że wirus, który ją powodował, wymarł. Dlaczego więc o SARS już tyle się nie mówi? Po wielkich alarmach z roku 2003, kiedy to zaczęto mówić — a mogło być inaczej? — o nowej hiszpańskiej grypie i przez wiele miesięcy rujnowano handel i turystykę, choroba, o której pierwotnie tak było głośno, poszła w zapomnienie. (...) Tajemnica, jaką otoczona jest ta choroba, nie została rozwiana: nie jest jasne, jak dochodzi do zarażenia, jaka jest natura wirusa i dlaczego wirus zginął tak niespodziewanie, jak się pojawił." [ 72 ]
Daleki jestem od wywoływania paniki, zarazem pokaźny dystans dzieli mnie od uspokajania opinii publicznej. Choroba niespodziewanie może przybrać groźniejszą, nie znaną dotychczas postać, a jej konsekwencje ekonomiczne mogą się okazać druzgocące dla systemów gospodarczych Europy.
Opinia społeczna ulega łatwo manipulacjom, to smutny fakt, bezlitośnie wykorzystywany. Niemalże na zawołanie, z rozbrajającą łatwością ogniskuje się uwagę mas na SARS czy wściekliźnie. Nie trzeba ulegać manii, czy należeć do zwolenników rozmaitych spiskowych teorii dziejów, by nie zastanowić się nad milczeniem rządów na temat prionów. Przyznanie się do niewiedzy jest niebezpieczne, rodzi wiele niewiadomych. Ludzie zapomnieli, że jednym z podstawowych zagrożeń zdrowotnych planety pozostają malaria, gruźlica, AIDS. Choroby te bardzo słabo zaznaczają swoją obecność w mediach, zwłaszcza państw rozwiniętych. Dlaczego?
Poziom wiedzy realnej dramatycznie spada, zamieniamy się w konsumentów rzeczywistości. Ponieważ jej nie rozumiemy wymagamy, aby inni, kompetentni zaoferowali nam gotowe wyjaśnienia, wzorce postępowania. Do biologicznych czynników chorobotwórczych zaliczono: pasożyty, stawonogi (kleszcze, komar, muchy tse-tse i in.), priony, mikroorganizmy patogenne (głównie bakterie), wirusy. Czytelniku, potrafisz wymienić konkretne cechy np. bakterii, wirusów. Czy wiesz, że antybiotyki nie działają na choroby wirusowe? Czy wiesz jaka jest tego przyczyna? Ginie krytyczna analiza informacji. Gorzką refleksję snuje Felipe Fernández-Armesto: „(...) "natarczywość informacji" upoiła umysły i prawdopodobnie otumaniła pokolenie (...)." [ 73 ]
Wracając do prionów przytoczę słowa Carletona Gajduska z lipca 1996 r.: "Nie mają najmniejszego pojęcia, co powoduje zachorowania wśród ludzi. To kuru i nic innego, tylko kuru, a nosicielem może być każdy gatunek — mleczne krowy, bydło hodowane na mięso, świnie, kurczęta. Powinni zdawać sobie sprawę z zagrożenia i podejść do tego realistycznie. Wszystkie świnie w Anglii karmi się preparatem mięsno-kostnym. Choroba nie pojawiła się u świń, bo nikt nie trzyma ich przez siedem lub osiem lat; idą na rzeź najpóźniej w drugim lub trzecim roku życia. Kiedy wszczepiliśmy chorobę świniom w laboratorium, zapadały na scrapie po ośmiu latach. Prawdopodobnie wszystkie świnie w Anglii są zakażone. A to oznacza, że nie tylko ich mięso jest niebezpieczne. Niebezpieczny jest także (...) portfel ze świńskiej skóry i katgut chirurgiczny wytwarzany ze świńskiej tkanki. Wszystkie kurczaki karmi się preparatami mięsno-kostnymi i one prawdopodobnie także są zakażone. (...) Wegetarianie mogą się zarazić, bo przy uprawie jeżyn stosuje się kurzy nawóz. To może być w tłuszczu, w maśle — jak do diabła mam określić zakaźność masła? Nikt na świecie nie ma pojęcia, jak to zrobić. Chorzy na CJD byli krwiodawcami. Tak więc we krwi również może być czynnik zakaźny. (...) to może być także w mleku. Przecież tego nie wykluczono."
Gajdusek może się mylić i zaklinam los, by rzeczywiście tak było. Bulwersuje mnie jednak oraz niepokoi brak nagłośnionych, ponadnarodowych akcji profilaktycznych i edukacyjnych związanych z prionami. Czy przy tej liczbie znaków zapytania ludzkość stać na tak mariaż ignorancji z arogancją?
Tylko dobrze wyedukowane społeczeństwo daje nadzieję na szybkie, skuteczne reagowanie w sytuacjach kryzysowych. Dekadę temu Europę Zachodnią ogarnęła panika. Od października 2000 r. spożycie wołowiny w Niemczech spadło o 50 procent; podobnie jest w Hiszpanii, Włoszech, Grecji i Francji, spadek konsumpcji szacowano tam ponad 40 procent. W Wielkiej Brytanii po informacjach naukowców, że co roku na nową odmianę choroby Creutzfeldta i Jakoba może zapadać 5-200 tys. osób, wycofano wołowinę ze stołówek w 10 tys. szkół, a sprzedaż tego mięsa gwałtownie zmalała. Ceny wołowiny w całej UE spadły o 27 procent, co w praktyce oznacza załamanie rynku. Zbliżony scenariusz pozostaje realnym.
Nie wiadomo, ile osób zakaziło się BSE przed lipcem 1988 r.
Ostatnie doniesienia z ostatnich lat:
Naukowcy z Whitehead Institute w Cambridge (Massachusetts) prowadzą badania mające na celu produkcję z prionów supercienkich drutów. Prionowe włókna można połączyć z cząsteczkami złota, co umożliwia przewodzenie prądu. Druciki dzięki cechom prionów są wytrzymałe, odporne na gotowanie oraz działanie rozpuszczalników organicznych.
W czasopiśmie „The Journal of Infectious Diseases" przedstawiono wyniki badań nad enzymem bakteryjnym kreatynazą, który może pomóc w zwalczeniu prionów. Na razie badania zakończyły się sukcesem w warunkach in vitro. Priony poddane działaniu kreatynazy oraz detergentu ulegały rozkładowi. Trwają badania mające na celu określenie czy kreatynaza może rozkładać priony w organizmie zwierzęcym/ludzkim. (styczeń 2004 r.)
Szczepionkę przeciw prionom przygotowali naukowcy z Uniwersytetu w Nowym Jorku, pod kierownictwem prof. Thomasa Wiśniewskiego. Pozwoliła ona znacząco opóźnić rozwój choroby wywołanej przez priony, podobnej w objawach do BSE; 30 procent zaszczepionych a następnie zainfekowanych osobników myszy, przeżyło ponad 500 dni — nie zaszczepione żyły przeciętnie 185 dni, maksymalnie do 300. (maj 2005). W przyszłości być może do otrzymania zwierząt niepodatnych na zakażenie prionami zaprzęgnięte zostaną techniki inżynierii genetycznej.
Na przełomie lat 2009/2010 zespół dr. Charlesa Weissmanna opublikował materiały w których wykazywano po raz pierwszy w historii możliwość zajścia u prionów procesu ewolucji, niespotykanego wcześniej u białkowych form zakaźnych. Naukowcy z Scripps Research Institute, twierdzą, że priony mogą podlegać ewolucji i selekcji naturalnej. Pierwszy etap przeprowadzonego przez nich eksperymentu polegał na przeniesieniu prionów z mózgów do komórek hodowanych in vitro. Po pewnym czasie populacja patologicznych białek szybko zmieniła swoją strukturę i została zdominowana przez formę PrPSC dostosowaną do rozwoju w hodowli komórkowej. Po przeszczepieniu patologicznego białka z powrotem do mózgów sytuacja odwróciła się, a większość w populacji znów zaczęły stanowić priony zoptymalizowane do wzrostu in vivo. W dalszej części doświadczenia ponownie pobrano priony z mózgów i umieszczono je w hodowli komórek nerwowych. Tym razem jednak, po osiągnięciu dominacji przez „hodowlaną" formę PrPSC, do pożywki dodano swainsoninę — substancję występującą u niektórych roślin i grzybów, znaną ze swojej zdolności do spowalniania rozwoju infekcji prionowej. Efektem interwencji było szybkie wytworzenie formy prionu opornej na działanie tego środka. Po dodaniu pożywki niezawierającej swainsoniny ponownie stwierdzono wzrost odsetka prionów wrażliwych na lek. Praca jest pierwszym dowodem na istnienie selekcji naturalnej oraz ewolucji wśród prionów. Dokonane odkrycie może oznaczać, że próby leczenia chorób prionowych powinny skupiać się nie na zmieniających się nieustannie formach PrPSC, lecz na PrPC, zachowującej wysoką jednorodność dzięki niezmiennemu „przepisowi" na to białko zapisanemu w genach.
Główne materiały źródłowe:
- Program multimedialny, Biologia komórki, Prószyński i S-ka S.A.
- „Wiedza i Życie" (numery: czerwiec 1996, lipiec 1996, styczeń 2000, marzec 2000)
- Rodzinna encyklopedia zdrowia, Świat Książki & Wydawnictwo Lekarskie PZWL, 1999, t. III.
- Praca pod red. Otałęga Z., Encyklopedia biologiczna, Agencja Publicystyczno-Wydawnicza Opres, Kraków, 1998-1999, t. II, VIII.
- Gulisano P., Pandemie. Od dżumy do ptasiej grypy, Wydawnictwo Polskiej Prowincji Dominikanów W drodze, Poznań 2007, ss.177.
- Rhodes R., Śmiertelne uczty. Tropem tajemnic nowej groźnej epidemii, Wydawnictwo „Książka i Wiedza", Warszawa 2000, ss.214.
- „Epidemia strachu" [w:] „Wprost" numer: 7/2001 (951)
- 18 grudnia 2000 r. — notatki z nagrania dokonanego na wykładzie zorganizowanym przez Uniwersytet Warszawski i Wydawnictwo Prószyński i S-ka (w ramach tzw. kawiarenki naukowej).
- 15 stycznia 2001 r. — aktualizacja [za:] „Gazeta Wyborcza" (artykuł autorstwa Artura Włodarskiego)
- 13 kwietnia 2001 r. — aktualizacja m.in. w oparciu o internetowy dodatek naukowy „Gazety Wyborczej" z marca 2001 r.
- 22 maja 2001 r. — wycinki prasowe
- Grzeszkowiak W. „Odkryto ewolucję u prionów" w:] http://kopalniawiedzy.pl/
- Ishikawa T. „Priony — u ssaków i drożdży" [w:] http://www.olimpbiol.uw.edu.pl/UserFiles/File/wyklady/priony.pdf
- Zużewicz M. A. „Priony — zagrożenia biologiczne spuścizną byłego tysiąclecia", Akademia Medyczna w Warszawie Katedra i Zakład Mikrobiologii Lekarskiej, 2001 r. [w:]http://www.ciop.pl/7159
- http://pl.shvoong.com/exact-sciences/
- Materiały Koła Naukowego Neurologii przy Klinice Neurologii Dorosłych Akademii Medycznej w Gdańsku [w:] http://kolo-neuro.gumed.edu.pl/
- http://www.przyroda.cad.pl/
1. z pojedynczą nicią o dodatniej polaryzacji ssRNA(+)
2. z pojedynczą nicią o ujemnej polaryzacji ssRNA(-)
3. o podwójnej nici dsRNA (grupa ta obejmuje reowirusy).
Poza tym podziałem znajdują się retrowirusy (np. HIV, wirus grypy), zawierające dwie nici RNA o dodatniej polaryzacji; nie są one jednak komplementarne jak u reowirusów, ale identyczne i tylko częściowo połączone. Materiał genetyczny wirusów o +RNA jest zakaźny (z wyjątkiem retrowirusów, wymagających do replikacji dodatkowo odwrotnej transkryptazy).
1. model polimeryzacji - zakłada tworzenie się ziaren - ośrodków agregacji powodujących zmianę konformacji kolejnych cząstek PrPc hipoteza szablon-pomocnik (template-assistance) /hipoteza heterodimerów - PrPc wiąże się z PrPSc lub nieprawidłowo sfałdowanym białkiem prionowym (heterodimer), po czym ulega przekształceniu do PrPSc - powstaje homodimer, który rozłącza się, aby utworzyć kolejne heterodimery.
Centralnym laboratorium diagnozującym tę chorobę w Polsce, mającym akredytację Unii Europejskiej jest Państwowy Instytut Weterynaryjny w Puławach. Badania mogą być również wykonywane w kilku Zakładach Higieny Weterynaryjnej. Materiał do badania jest pobierany z pnia mózgu padłych bądź poddanych ubojowi przeżuwaczy. Obowiązkowi badania podlegają padłe lub zabite owce i kozy powyżej 18 miesiąca życia oraz bydło powyżej 24 miesięcy.
- tzw. grypa sezonowa - zależności od sezonu epidemicznego charakteryzuje się śmiertelnością0,1-0,5 % (tzn. że umiera 1-5 na 1000 osób, które zachorowały).
- grypa pandemiczna A/H1N1v - w różnych regionach świata śmiertelność jest różna, a wartość zależy od sposobu oszacowania: w Europie waha się w granicach 0,1-1 %, w Ameryce Południowej sięga 0,7 % lub 1 %, a w Stanach Zjednoczonych 0,5-1 %.
- wśród chorych na grypę pandemiczną wymagających hospitalizacji i leczonych lekami przeciwwirusowymi umiera 7 %, ale pośród tych przyjmowanych na oddział intensywnej terapii śmiertelność waha się w przedziale 14-25 %. Bez leczenia przeciwwirusowego u chorych wymagających hospitalizacji śmiertelność może wynosić nawet ponad 41 %. [za:] http://www.mp.pl/grypa/
Krzysztof Pochwicki Nauczyciel, publikował w piśmie "Gameranking", współpracuje z miesięcznikiem "21. Wiek" (członek zespołu redakcyjnego). Autor książki "Cywilizacja traw". Pióro do wynajęcia. Nauczyciel, publikował w piśmie "Gameranking", współpracuje z miesięcznikiem "21. Wiek" (członek zespołu redakcyjnego). Autor książki "Cywilizacja traw". Pióro do wynajęcia.
Liczba tekstów na portalu: 14 Pokaż inne teksty autora |
Oryginał.. (http://www.racjonalista.pl/kk.php/s,951)
(Ostatnia zmiana: 25-02-2011)